Fenilcetonuria (PKU)
¿Qué es la fenilcetonuria?
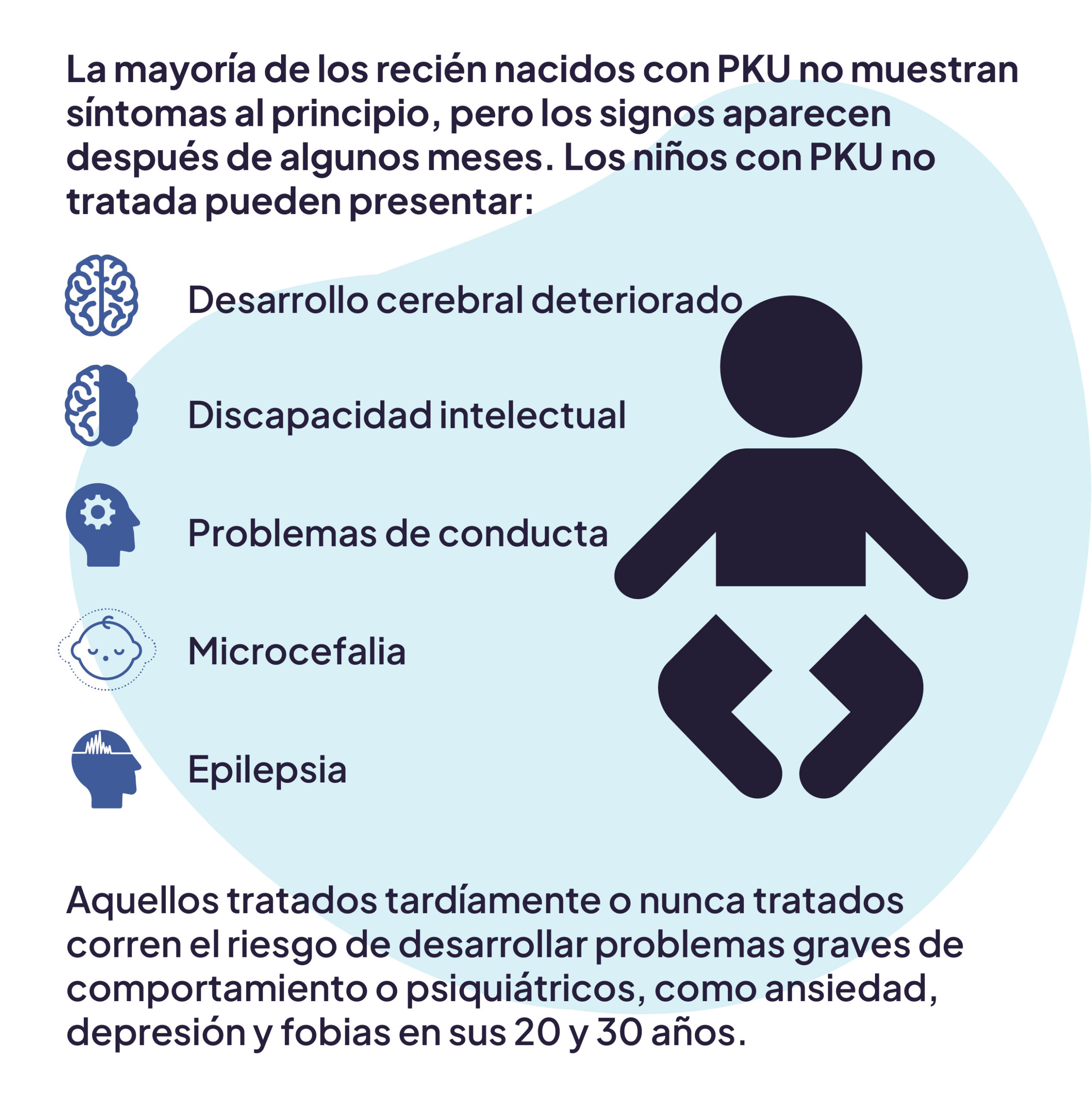
La fenilcetonuria (PKU) es una enfermedad metabólica hereditaria poco común que afecta el cerebro.1 Es causada por un defecto en el gen que ayuda a crear la enzima necesaria para descomponer la fenilalanina.1 Si no se trata o se maneja de forma inadecuada, la fenilalanina —un aminoácido esencial presente en todas las proteínas y en la mayoría de los alimentos— puede acumularse a niveles dañinos en el organismo. Esto provoca discapacidades graves e irreversibles, como discapacidad intelectual permanente, convulsiones, retraso en el desarrollo, pérdida de memoria y problemas conductuales y emocionales.1
Los recién nacidos con fenilcetonuria inicialmente no presentan síntomas, pero estos suelen aparecer progresivamente. El daño causado por niveles tóxicos de fenilalanina en los primeros años de vida es irreversible.2,3
El diagnóstico de fenilcetonuria generalmente se realiza mediante programas de cribado neonatal.4
¿Qué tan común es la fenilcetonuria?
Se estima que hay unas 58,000 personas afectadas en todo el mundo.
Hombres y mujeres tienen la misma probabilidad de heredar el gen defectuoso y desarrollar PKU.

¿Cómo está trabajando PTC para tratar la fenilcetonuria?
PTC está desarrollando un tratamiento potencial para la fenilcetonuria basado en nuestra plataforma metabólica.
Neil Smith Líder Global del Proyecto PKU en PTCSabemos por nuestra investigación que existe una gran comunidad de pacientes con PKU que necesita más opciones de tratamiento. También hemos construido relaciones increíbles dentro de esta comunidad, incluyendo pacientes y organizaciones de defensa. Ayudar a esta comunidad es mi misión y motivación, y mi confianza en la ciencia me ayuda a mantenerme enfocado y seguir adelante.
Do you have questions?
Please reach out if you would like to speak with us.
[1] de Groot MJ, Hoeksma M, Blau N, et al. Mol Genet Metab 2010;99:S86–S89.
[2] Phenylketonuria (PKU). Available at: https://www.mayoclinic.org/diseases-conditions/phenylketonuria/symptoms-causes/syc-20376302. Accessed October 2021.
[3] Blau N, van Spronsen FJ, Levy HL. Lancet 2010;376:1417–1427.
[4] Al Hafid N, Christodoulou J. Transl Pediatr 2015;4(4):304–317.